Sam Friedman is a Machine Learning Scientist in the Data Sciences Platform at the Broad Institute. He began working on variant calling and filtration over 5 years ago, bringing cutting edge tools from computer vision to bear on problems in genomics. Currently, he serves as tech lead for the Broad’s Machine Learning for Health initiative.
The Broad Institute’s Genome Analysis Toolkit (GATK) has long been an industry-leading source of tools and pipelines for variant calling. In particular, the GATK Best Practices pipeline for short variant analysis is heavily used worldwide for scalable analysis of Whole Genome Sequencing (WGS) data. However, that pipeline is optimized for co-analyzing samples from more than one subject, yet sometimes researchers need to analyze a single genome sample. In that case, the established methods that use Bayesian statistics don’t do quite as well on the filtering side as more recent algorithms like deep learning. That is why a few years ago, we developed a deep learning method for filtering short variants called GATK CNN (for Convolutional Neural Network) that performs better on single genomes than our cohort-based tools.
Over the past year, we found that these tools had reached the stage of development maturity where the quality of the scientific results they produce is consistently excellent, so we shifted our efforts to making sure the tools are easily maintainable and fit for production environments.
A key aspect of productionizing computational tools like these, which typically get run on high volumes of data, is to optimize their operation to run as quickly as possible. To that end, we’ve been collaborating with a team at Intel who developed an open-source toolkit called OpenVINO™ for speeding up machine learning algorithms built on the TensorFlow framework.
We wanted to see if using OpenVINO™ could help improve the runtime of the GATK CNN tools on Intel processors, starting with CNNScoreVariants, a GATK tool designed to apply a previously trained CNN model to a single-sample whole genome variant callset in VCF format. The output of the tool is a new VCF with variant quality annotations that can be used to filter the callset, thereby increasing its accuracy. Under the hood, CNNScoreVariants uses TensorFlow to compute the variant quality annotations.
Integration of OpenVINO™ easily delivered significant acceleration
We were able to hook up the CNNScoreVariants tool to use OpenVINO™ with just two lines of code, and found that in terms of variant throughput, it sped up operation by 21% for a run on a subset of a single 30x WGS sample containing 31,742 variants, compared to a baseline run of the original tool on the same sample (both runs done on a 4-core Intel i5 processor with 8GB RAM). In terms of wall-clock time, this reduced the runtime from 8.67 minutes to 7.6 minutes for our test callset, and would translate to a savings of approximately 4 out of 22.5 hours on an entire genome’s worth of data (~5 million variants). Not bad considering we didn’t even have to modify any of the tool’s existing code!
With help from our Intel collaborators, we also identified some functional tweaks that could accelerate the tool’s operation further. We modified some of the existing code to use a “frozen” model approach, leveraging functions that are available in OpenVINO™, and found that the number of variants processed per second increased even more with this approach. Specifically, it reduced the test sample runtime to 7.16 minutes, which amounts to shaving off another 1.5 hours of processing on a whole genome.
Taken together, these straightforward modifications resulted in an increase of 32% in the tool’s variant throughput compared to the baseline.
We also ran this on the cloud in Terra, and found a similar pattern of acceleration (~20%). There is variability in performance which can be expected with different environments; your results will also vary if you use this on your own computing infrastructure.
You can read more about the integration work and benchmarking results in the Intel team’s blog here.
Try it out in Terra
We wanted to make it easy for anyone to try this out in practice, so we set up a preconfigured cloud environment in Terra that has all necessary dependencies for OpenVINO™ and the relevant GATK tools already installed, as well as a Jupyter notebook set up to run the CNNScoreVariants tool with and without the OpenVINO™ optimizations.
To try it out for yourself, spin up a copy of the “OpenVINO integration with TensorFlow” environment, which is available from the Community-maintained Jupyter environments section of the custom cloud environments menu in the Terra app, shown below. Set the number of CPUs to 4 and the memory to 15 Gb.
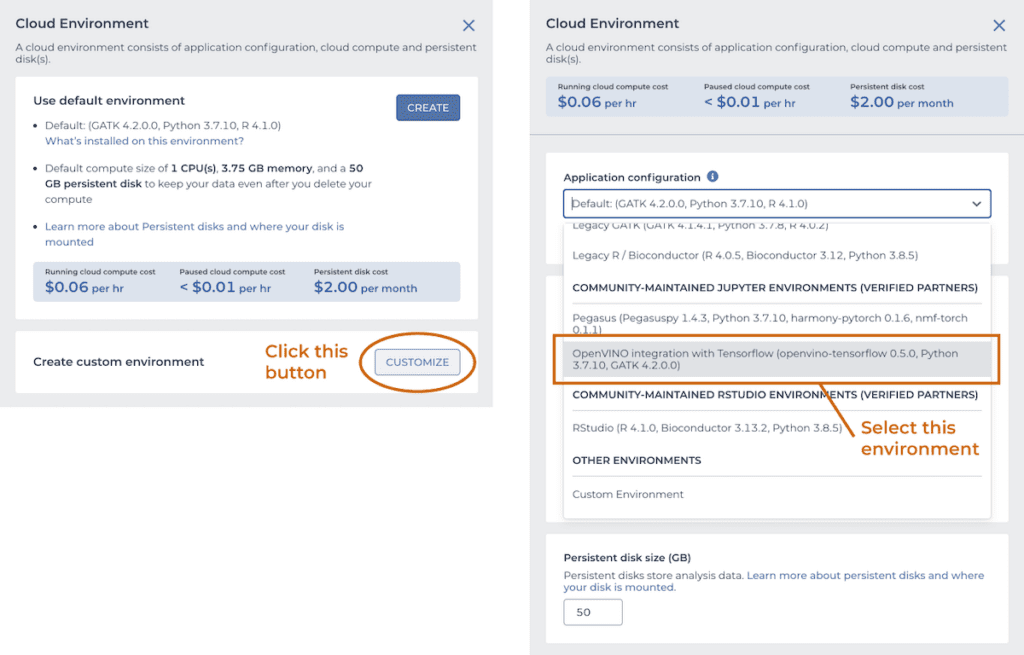
While the system spins up your OpenVINO™ cloud environment, import the GATK-OVTF-Notebook.ipynb notebook into a workspace. Once the system notifies you that your environment is ready, open the notebook in EDIT mode, and run the two GATK CNNScoreVariants commands. The last few lines of the log show how many variants were processed and how much time the run took.
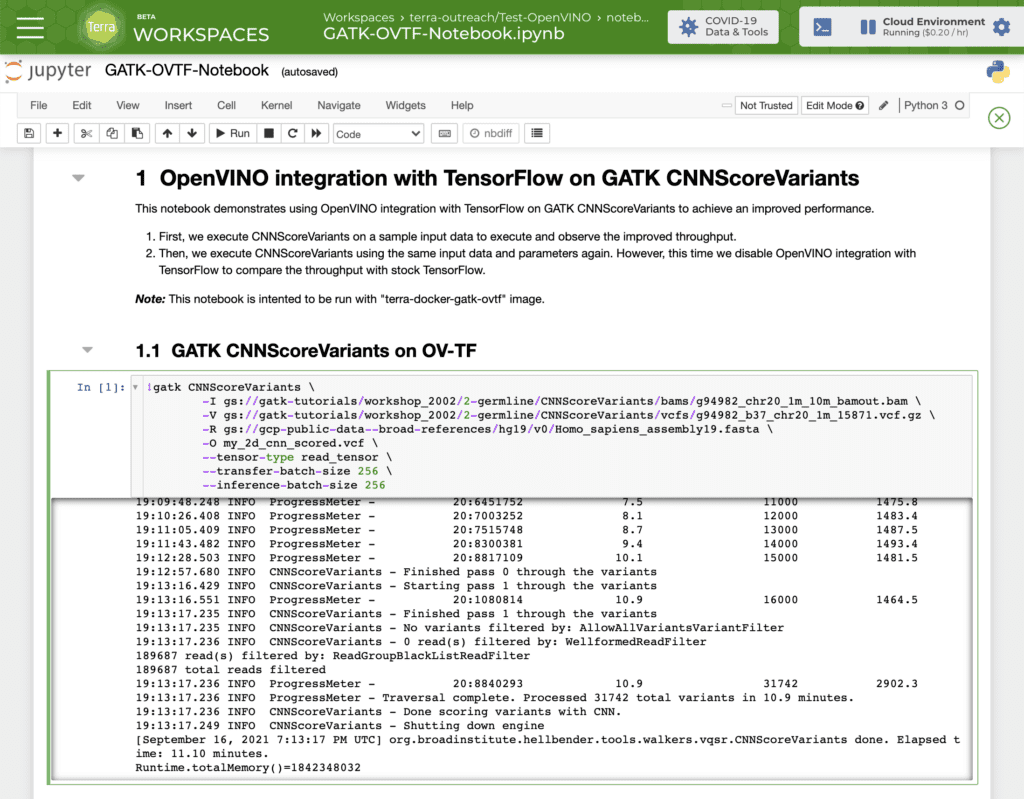
You can test these commands on your own data by replacing the paths to the input files, which must be located in a Google Cloud Storage bucket for which you have read access. If you don’t already have your data in a GCS bucket, you can drag and drop the files into the workspace’s dedicated storage bucket. You can also increase the number of CPUs allocated to your cloud environment if you want to test how performance scales according to compute resources.
If you’re new to Terra, have a look at the documentation for more detailed instructions on working with custom cloud environments and Terra workspaces.
Whether you’re a researcher using CNNScoreVariants in your genomic analysis work, or a developer interested in applying similar solutions to your own tools, we hope this behind-the-scenes peek at how we used OpenVINO™ integration with TensorFlow* to accelerate our tools will be helpful.
If you run into any trouble or have questions, please consult either the Terra Help resources or Intel’s OpenVINO™ support page, or submit your questions, feature requests and bug reports regarding OpenVINO™ via GitHub issues.
