Kylee Degatano is a Senior Product Manager in the Data Sciences Platform at the Broad Institute. In this guest blog post, she introduces the Genomic Variant Store, a highly scalable solution for genomic analysis based on Google BigQuery, designed to scale joint variant discovery to a million whole genome samples. Researchers interested in trying out this approach are invited to join the GVS Early Access program.
Researchers around the world are generating petabytes of genomics data to understand human biology, identify the causes of diseases and develop new treatments. The analyses involved, such as association studies, linkage analysis, and exploration of Mendelian inheritance all require high confidence in the quality of the genomic variant calls they rely on.
One of the most successful approaches of the past decade for generating such high quality variant calls involves jointly analyzing the genomes of many different people across a population. This “joint calling” approach increases statistical power for differentiating true variants from artifacts, which makes it possible to identify extremely rare variants with confidence. In the GATK Best Practices for germline short variant discovery, it is implemented as a two-step process: first we identify potential variants individually per sample, then we evaluate the evidence found for each genomic site across all samples to produce “joint calls”, i.e. a multi-sample variant callset. We can then apply additional filtering to further refine the callset for downstream analysis.
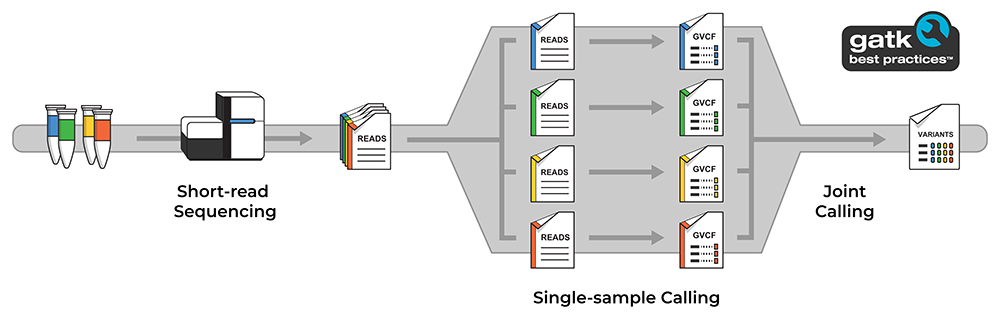
Diagram illustrating the overall data generation and analysis process involved in joint variant discovery.
As an example of how this approach empowers discovery, my colleague Laura Gauthier recently wrote a blog post commenting on a recent study that used a joint callset of 75,000 exome samples to implicate ten new genes in the development of schizophrenia. Crucially, she explained, the study focused on ultra-rare variants, never seen before and only observed in a single individual in that entire callset, that affected the same small set of genes. The joint calling methodology was essential to the success of that analysis because it enabled the authors to accurately detect ultra-rare variants with confidence, and to do so for enough individuals to accumulate enough statistical power to implicate the corresponding genes. Laura concluded her post with this prediction:
“Future breakthroughs in common disease research will continue to come about through the hard work of recruiting large numbers of affected participants; accurately detecting the few, tiny, rare mutations that make them different; and combining as many ultra-rare variants as we can until we can point the finger at genes harboring too many mutations across people who suffer from disease.”
It is with that same vision in mind that our engineering team has been working hard to scale up our joint calling capabilities.
Every jump in the scale of the studies we have supported so far has required that we re-engineer various parts of our analysis pipelines to address new barriers related to cost efficiency, computing power, runtime… or the maximum amount of data you can store in a single file on a standard hard drive. (That was a fun one to hit.)
Today, I’m excited to share the solution we developed to get to the next order of magnitude, and to make this kind of scaling accessible to a wider range of researchers and institutions.
Aiming for a million
A few years ago, the NIH asked our team to figure out a way to run joint calling on one million human genomes for the All of Us Research Program. For context, at the time, performing joint calling for “just” 15,000 whole genomes was a costly months-long endeavor for a full team of engineers equipped with cutting-edge tools. We knew that to scale to a million genomes, we would need to revisit the engineering design behind the GATK Joint Calling pipeline (again). We also foresaw that many downstream analysis tools would not be able to handle the resulting callset, which would contain trillions of variants. Researchers would need to be able to subset the variant calls to their samples and genomic regions of interest.
Oh, and we needed to ensure our solution to these problems would be cost- and time-efficient, and scalable to both ends of the spectrum: it should allow us to scale down to 10 samples as easily as scaling up to a million.
So we took joint calling back to the drawing board. We started by designing a core schema for the variant data based on data access patterns for key use cases like training a filtering model, searching to identify samples with a specific variant or variation in a specific location, and extracting data —for all samples or for subsets— into Variant Call Format (VCF). We contemplated which information in the variant files was necessary for joint calling and downstream analysis, and stripped out anything extraneous. We tested many different foundational technologies for processing and storing data, including Spark, Dataflow, and custom developed infrastructure. We ultimately chose Google BigQuery because it is easy to operate, can handle huge data sizes, is cheap to store data and cheap to query, and has excellent security features.
The result of this three-year development effort: a highly scalable and cost-effective variant storage and processing solution we call the Genomic Variant Store.
Introducing Joint Calling with the Genomic Variant Store
The Genomic Variant Store (GVS) uses Google BigQuery to store variant data according to the core data model. It is designed to function as a persistent data warehouse to which we can add new data over time, simply by importing gVCF files produced by the single-sample calling step of the variant discovery process outlined above. This automatically combines the per-sample variant data across samples and genomic coordinates. We can then query the GVS by sample and/or by genomic coordinates to generate subset callsets of interest in either VCF or Hail VDS file format.
If you are familiar with the details of the current GATK Best Practices workflow implementation, the GVS data ingestion step is analogous to combining gVCFs into a GenomicsDB data store. However GVS is much more scalable than GenomicsDB due to its use of Google BigQuery.
The GVS also includes a built-in variant filtering model (GATK VQSR) that determines which variant calls will be considered true variants, as opposed to artifacts. The filtering model is applied when a subset of variant data is extracted to file, and it can be re-trained and improved as new data is added.
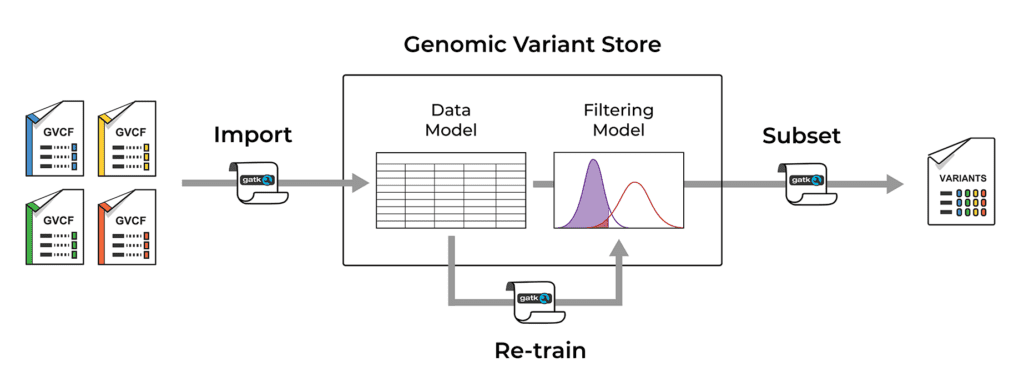
Overview of the operations supported by the Genomic Variant Store
For convenience, we have developed utility workflows written in the Workflow Description Language (WDL) to perform the import, model training, subsetting and variant search operations. These can be used individually to grow, curate and analyze a persistent variant data store.
We have tested this on a wide range of callset sizes; there is no minimum number of samples, and we anticipate that it will scale to the planned one million genomes for the All of Us Research Program. In fact, we have already used the GVS in production to produce a joint callset from 250,000 human whole genomes for the AoU program, which we believe is the largest joint-called human whole genome callset in the world so far.
Get early access to a self-contained GVS Joint Calling Pipeline
Due to the engineering challenges involved in operating at this scale, there are only a handful of genome centers around the world that are currently able to create joint callsets from tens of thousands of human whole genomes, let alone hundreds of thousands. Yet it is very likely that progress in human genetics could be substantially accelerated if this capability were made more widely accessible.
As we continue to improve the GVS — increasing scalability, adding support for new data types, improving the internal algorithms — we are looking for feedback from external groups to ensure the GVS will work well for a wide audience. To that end, we are starting an early access program for researchers who are interested in trying out the GVS approach for making joint callsets of up to 10,000 human whole genomes.
We are currently targeting that project size because most population genetics projects today involve less extreme cohorts than the All of Us Research Program; we’re seeing many projects with whole genome sample numbers in the low thousands.
To enable those projects to use GVS for scalable joint calling without having to manage complex infrastructure and run multiple separate operations, we developed a “one and done” workflow called the GVS Joint Calling pipeline that wraps all the necessary steps. This single self-contained workflow takes in a set of per-sample gVCFs, trains and applies the GATK VQSR filtering model, and extracts the variants into a VCF file containing the complete joint callset.
We plan to make this pipeline publicly available in a Terra workspace, pre-configured in such a way that anyone can run it out of the box with minimal effort. In our current tests, the GVS Joint Calling pipeline set up in Terra can produce a joint callset from up to 10,000 human whole genomes in less than half a day, at a cost of $0.06 USD per genome.
Screenshot of the Terra workflow configuration panel for the GVS Joint Calling Pipeline
We invite you to apply to join the early access program by filling out a short form that will help us assess if your callset would be a good fit for the initial release. If you are selected, we will help you get started with the GVS Joint Calling pipeline in Terra, and we will be available to assist you if you run into any problems.
Even if the Genomic Variant Store doesn’t meet your needs right now, please feel free to use the form to tell us more about your work and what features you be interested in seeing in a future version. We look forward to hearing from you!
