Laura Gauthier is the Director of Germline Computational Methods in the Data Sciences Platform at the Broad Institute. As a computational biologist and longtime GATK team member, she has both personally contributed to and overseen the development of key algorithms and tools for genomic analysis. In this guest blog post, she provides an inside perspective on the methodologies underpinning recent discoveries in common disease research.
We recently witnessed what’s been called a “watershed moment” of schizophrenia research, marked by the back-to-back publication of two major studies that reveal genes and genome regions that influence schizophrenia risk, as summarized in a Broad Institute blog post. One of the studies, a large meta-analysis done by computational biologist TJ Singh and colleagues, used a combination of several exome cohorts totalling about 75,000 samples to implicate ten new genes in the development of schizophrenia.
This study was a departure from many classic genome-wide association analyses of the past, which typically compared the frequencies of individual variants between the “case” and control groups. The variants in question were “common” (usually >1% of the population), but each had only a very small effect on its own. In contrast, the new study focused on ultra-rare variants that were each predicted to have a large biological effect on their own, but most of which were present in only one person in the study cohort. By aggregating these ultra-rare variants per gene to improve statistical power and comparing the number of deleterious mutations across whole genes, the authors were able to highlight several new genes with big implications for schizophrenia that hadn’t been found before using the common variant approach.
This discovery would not have been possible without the 75,000 sample callset, which, at the time these data were generated, was pushing the boundaries of what was computationally possible — though since then, gnomADv3 has doubled that number with a jaw-dropping 150,000 samples. It was also exactly what we had in mind as we worked to improve the accuracy and scale of the GATK joint calling pipeline.

Different paths to finding variants that cause disease
The most intuitive model of genetic disease is what we remember from high school biology: an unfortunate mutation in a single, important gene leads to a dysfunctional protein that impacts the affected individual’s biology. One example that might come to mind is sickle cell anemia, when a mutation in the red blood cell protein HBB allows cells to collapse or break apart and die, preventing them from effectively carrying oxygen and leading to painful blood vessel clogs.
For many rare diseases where the defective gene is not yet known, we can compare the genomes of family members who are affected and unaffected by the disease, and identify one or two variants (sometimes called mutations) that have a very large “effect size”, i.e. that cause most if not all of the symptoms that characterize the disease.
In contrast, schizophrenia and most psychiatric disorders fall under the category of “common disease”, which are typically caused by a constellation of many variants with much smaller effect sizes combining to cause disease. That requires us to apply a different approach to identify contributing variants: instead of studying a small set of family members, we compare large cohorts of cases (affected individuals) and controls (unaffected individuals) in studies that typically include thousands of participants.
The challenge of common disease genomic research is that population geneticists have observed an inverse relationship between the prevalence and severity of variants involved in common diseases: the more common the variant, the smaller its effect size; and conversely, variants with large effect sizes are rare. So while we can “easily” find common variants that explain a tiny portion of the overall condition, we need to recruit a huge number of patients to be able to find those rarer variants that play a bigger role.
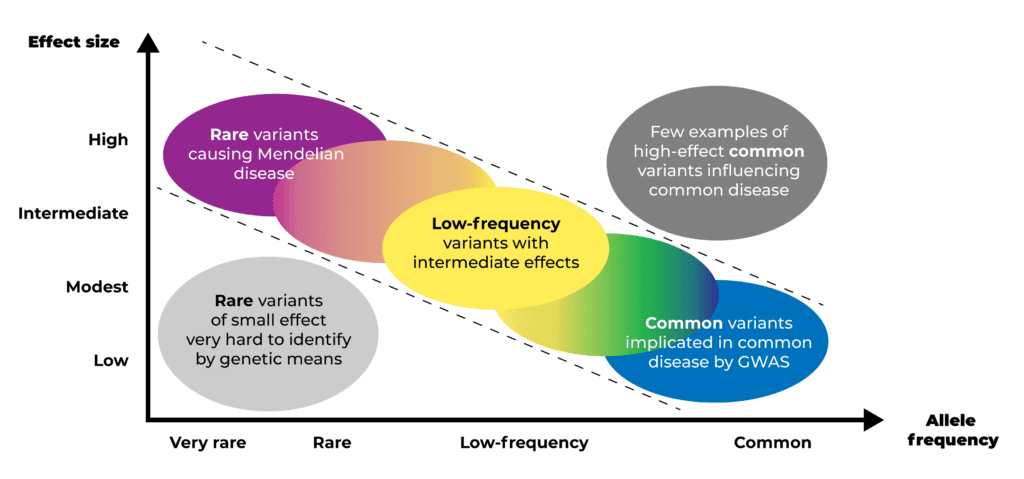
Diagram illustrating the inverse relationship between the prevalence and effect size of variants involved in common diseases. Derived from Manolio et al., 2009.
That is what makes this paper so groundbreaking: the study that revealed these new genes hinges on ultra-rare variants. The investigators had to accurately detect variants that have never been seen before and are observed in only one individual. And they had to do it for enough individuals to accumulate enough statistical power to implicate the corresponding genes.
Powering discovery through accuracy at scale
Finding ultra-rare variants is hard, mainly because when something is seen only once, it’s difficult to be sure it’s real and not just an artifact of the data generation process. In this study, the investigators used GATK tools to consider evidence from the entire cohort and evaluate individual variant calls with higher accuracy. Specifically, they used our GATK/Picard single-sample exome analysis pipeline to process data from each sample in a consistent way, and our GATK joint calling pipeline that can scale to tens of thousands of samples.
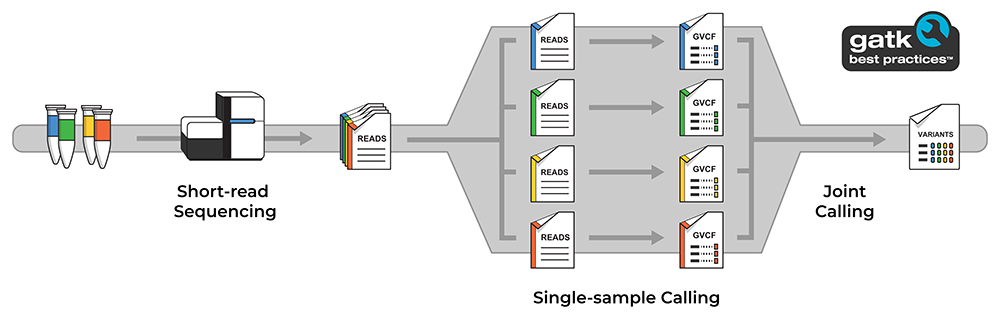
Diagram illustrating the overall data generation and analysis process involving the single sample and joint calling pipelines. See Resources below for links to relevant code and documentation.
This has been the team’s mandate since the GATK’s inception circa 2010. Originally embedded within the Broad’s Medical and Population Genetics (MPG) Program, the GATK team was created specifically with the mission of empowering researchers to perform accurate variant discovery from genome sequencing data. Later we left MPG to help form the Data Sciences Platform, but we continue to work closely with collaborators throughout the Broad and the larger genomics community in the pursuit of that exact same goal — and at increasingly larger scale.
Since 2010, we have been involved in many high-profile projects that have applied this approach successfully and we have seen countless others do so elsewhere on their own, enabled by our tools. It is intensely gratifying for us to see this work bear fruit once again, this time in the service of tackling such a complex condition as schizophrenia. But of course, the story doesn’t end with the list of new disease genes. Other scientists will take the new genomic results to guide in vitro experiments. In a related Broad Institute blog post, Stanley Center co-director Morgan Sheng explains how these results could potentially be actionable and lead to real changes in psychiatric disease treatment.
At the end of the day, achieving better accuracy isn’t about publishing papers or getting to the top of the leaderboard on the PrecisionFDA Challenge. Generating larger cohorts isn’t just about flexing our compute muscle. Future breakthroughs in common disease research will continue to come about through the hard work of recruiting large numbers of affected participants; accurately detecting the few, tiny, rare mutations that make them different; and combining as many ultra-rare variants as we can until we can point the finger at genes harboring too many mutations across people who suffer from disease.
Resources
The Picard/GATK pipelines referenced in this work are available as open-source workflows (written in WDL) from the Broad Institute’s WARP repository.
- Single-sample exome pipeline: code | pipeline documentation |
- Joint-calling exome pipeline: code | method description |
These workflows are also available in a public Terra workspace, preconfigured to run on example data. For more information on running workflows in Terra, see the Workflows Quickstart video tutorial on YouTube.
