Adam Nichols is a Senior Software Engineer in the Data Sciences Platform at the Broad Institute. He is a member of the Batch Workflows team, which is responsible for key capabilities related to launching WDL workflows in Terra. In this guest blog post, Adam takes us on a tour of the inner workings of the workflow execution system, highlighting recent changes the team made to reduce latency and improve user experience.
As one of the platform’s foundational features, Terra’s workflow execution system was designed to be incredibly robust and scalable. We pride ourselves on supporting ambitious projects such as the All of Us Research Program that require efficiency and consistency at enormous scale. Yet we also want to make sure that smaller research groups are able to get their work done smoothly as well, and that method developers who are scale-testing new workflows can iterate rapidly without being held up by unnecessary latency. This is a challenging balance to strike, and the range of specific needs we observe evolves over time, so we regularly revisit key components of the system.
In the first quarter of 2022, we cast a wide net to identify opportunities for improvements that would reduce latency and/or increase parallelism in the system. We found four specific areas where we were able to make highly impactful changes that result in a more consistent and responsive user experience, as I detail below. This work has also opened up some additional possibilities that we are going to follow up on in the coming months.
How it all works under the hood
Before I get into the nitty gritty of the changes we made, you need to know a few basic things about how Terra’s workflow system works. Briefly, there are three Terra subsystems involved: the User Interface (UI), an internal component called Rawls, and the Cromwell workflow engine; plus of course the cloud backend, i.e. Google Cloud Platform.
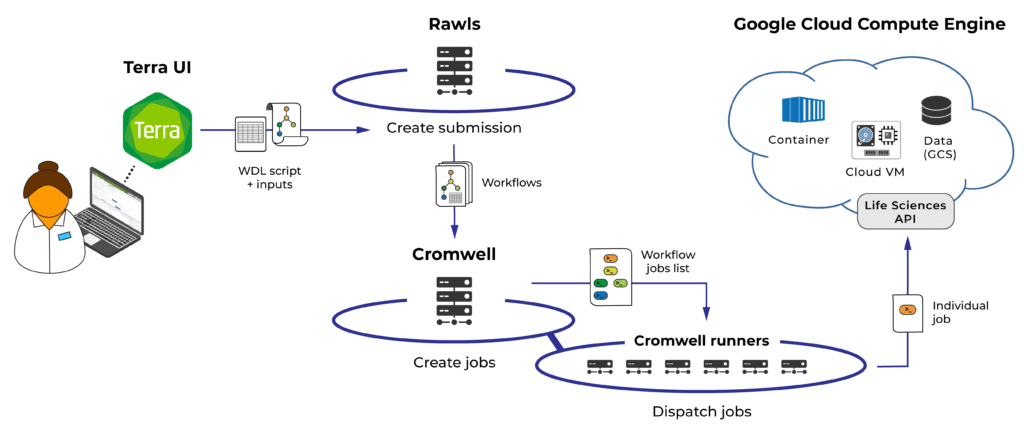
Overview of the workflow submission process (detailed description below)
To run a workflow in Terra via the UI, you start by specifying the input data you want to run it on, hit the “Run Analysis” button, then hit “Launch” in the confirmation menu. At that point, the UI sends instructions to the Rawls subsystem to create a “submission”, i.e. a collection of one or more workflow launch requests — one if you’re just feeding all the input data to a single instance of the workflow, more than one if you’re running the same workflow on multiple data entities (such as samples) separately.
Rawls then submits individual workflow launch requests to Cromwell, with their associated input definitions. Cromwell “picks up” workflows from the incoming queue and interprets their WDL code, traversing the specified tasks and defining the jobs (i.e. specific command lines) that need to be run on the cloud backend.
Cromwell finally submits these jobs to the Google Cloud Life Sciences API, which is responsible for provisioning Google Cloud Engine virtual machines (VMs) and executing jobs on those VMs. As jobs finish, Cromwell writes the corresponding logs and outputs to cloud storage; once all jobs in a workflow have completed, the workflow terminates. Rawls detects termination, updates the workflow’s status in the UI and registers the outputs in the relevant data tables. Finally, when all workflows in a submission have terminated, Rawls updates the submission’s overall status in the UI.
The overall process is subject to quotas in a few key places:
- Rawls supports up to 3000 concurrent workflows per user (across all submissions); any excess workflows are held in a user-specific queue.
- Cromwell uses a “constellation” of instances cooperating to perform various tasks, distribute load, and provide increased uptime with redundancy. Workflows execute on the “runner” instances, of which there are currently six; each runner instance allocates a quota of 4,800 jobs per billing project (which can be shared by multiple users).
- GCP imposes quota limitations on hardware resources and public IP addresses, which can be raised on request.
You might think that the most obvious way to make people happy would be to beef up system resources to allow for higher quotas. In fact, we found that just by tweaking how we pass requests from one service to another, we were able to effect major improvements to the user experience, without even touching any of the quotas currently in place.
Use batches to go from Submitted to Running in a fraction of the time
First we looked at how Rawls was submitting workflow orders to Cromwell: one at a time, resulting in a rate of around 125 workflows submitted per minute. That may not sound bad, but incoming submissions often contain hundreds to thousands of workflow launch requests; at that rate it would take 24 minutes to saturate the 3,000 workflow per user quota. This represents a lot of wall-time overhead, so we set ourselves the goal of dramatically improving the time it takes to reach 3,000 running workflows.
We immediately hit on the idea of having Rawls submit workflows to Cromwell in batches rather than one at a time, and that’s where we caught our first big break. Digging into the code, we realized that Rawls itself has always supported submitting workflows to Cromwell in batches. The batch size had just never been increased beyond 1. (We think this dates back to a much earlier phase of Terra’s development when there were a lot fewer people running workflows in the system.)
Unfortunately, we found out we couldn’t just go ahead and raise the batch size right away. Due to factors that we can’t control directly, the first few batches of a large submission sometimes fail seemingly randomly. Until now, we could tolerate that because the disruption was minimal — having to restart a couple of workflows is annoying, but not a showstopper — but setting a larger batch size would mean having to restart potentially hundreds of workflows in case of a random failure.
So we took a little detour to address the issue. We couldn’t fix the source of the problem, but we were able to engineer a new automated retry mechanism that handles the failure in the background. That in itself was an early win, since it meant you no longer have to manually relaunch workflows affected by this bug — the system just does it automatically on your behalf.
With that blocker out of the way, we tested a range of values for the batch size and found that a batch size of 50 achieved our goal very nicely, bringing the “time to saturation” down to just 35 seconds! This makes the biggest difference when running a large number of short workflows, because the time waiting to start could make up a significant amount of the total time. (Workflows beyond the first 3,000 start as quickly as the previous workflows finish and free up their quota slots.)
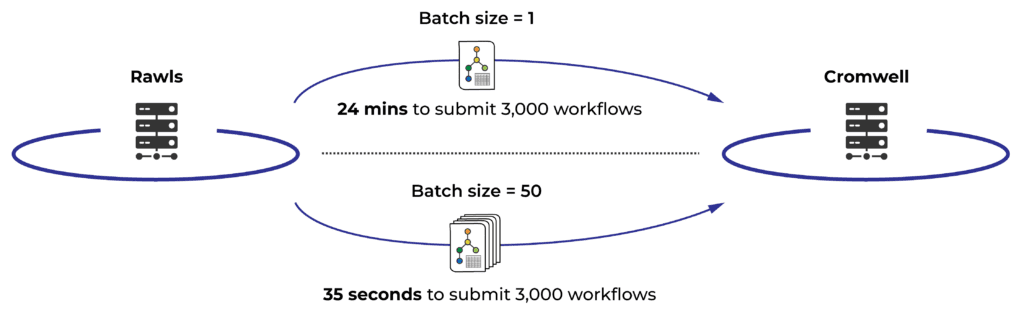
Dramatic improvement in submission time due to batching by Rawls
It’s rare that you can just change an integer in a configuration file and get a perfectly scaled outcome in the application, but that is exactly what happened here: workflows now submit in less than a minute.
Distribute workflows evenly to runners to max out concurrent jobs
Next, we looked at factors that might prevent workflows from progressing once they’ve been submitted to Cromwell.
As I mentioned earlier, Cromwell in Terra uses multiple “runner” instances for workflow execution. At present the system uses six such runners, and each of them allocates a quota of 4,800 jobs per billing project. That means Terra can submit up to 28,800 concurrent jobs on your behalf (per billing project) to the cloud backend — if you have workflows assigned to all six Cromwell runners.
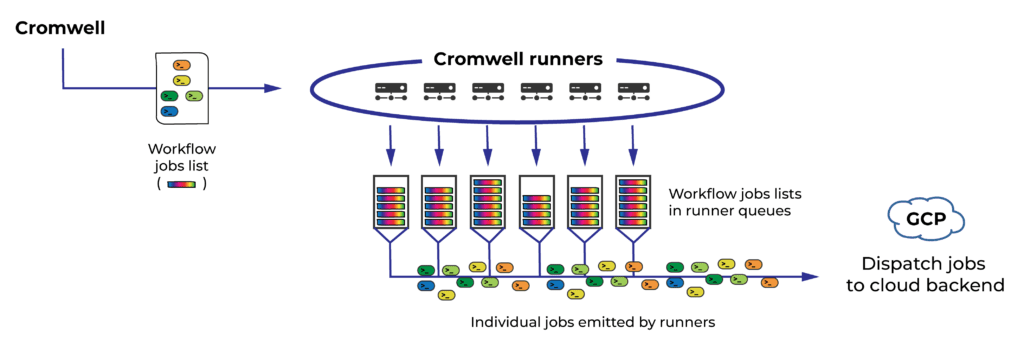
Cromwell runners pick up workflows (which at this points are collections of jobs to run) and dispatch individual jobs to the cloud backend for execution
So how do workflows get distributed to runners? Until now, runners picked up workflows in batches of 30. For submissions of hundreds or thousands of workflows, the resulting distribution ends up being relatively even across all runners, and overall this has worked well enough to enable people to make the most of their allocated capacity.
However, we eventually ran into a situation where someone submitted a small number of workflows that each generated many thousands of jobs. This unusual shape, combined with the non-uniform pickup algorithm, triggered a very uneven distribution of jobs across runners. The researcher quickly saturated their 4,800 jobs on one runner, while their allocations on the rest sat unused. This unnecessarily reduced the rate at which their workflows could be executed, making them look “stuck”, which made the researcher understandably anxious and unhappy.
The solution in this case was the reverse of how we dealt with Rawls: have the runners pick up only one single workflow at a time instead of 30, and count on the problem to solve itself statistically.
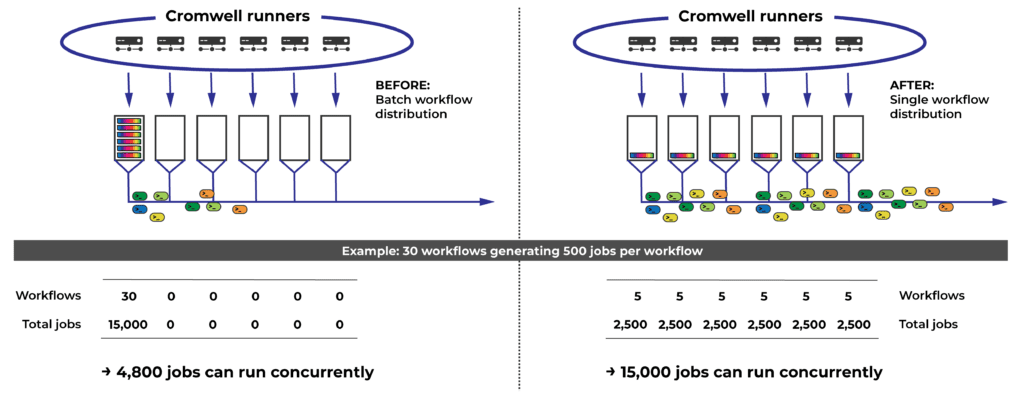
Getting rid of batching when distributing workflows to runners can dramatically increase the amount of concurrent jobs (for workflows that generate many jobs) given the cap of 4,800 concurrent jobs per billing project per runner.
Although the exact scale of the gains vary with workflow shape and cannot be generally quantified, we expect this improvement to especially benefit people who are running workflows that generate lots of concurrent jobs (for example, by using a lot of scatter parallelism).
Queue workflows explicitly when waiting for job capacity
Our work on the pickup algorithm used by the Cromwell runners (above) also gave us the opportunity to reevaluate how to handle incoming workflows when the job capacity for the corresponding project is already saturated.
Consider the million-job scenario: you submit 1,000 workflows, each of which launches 1,000 jobs. Because 1,000 workflows easily fit within the Rawls quota of 3,000 concurrent workflows, all of the workflows immediately start running in Cromwell, which proceeds to launch 6 x 4,800 = 28,800 jobs across the six runners. That job capacity is enough for 29 of the 1,000 workflows to make progress, while the remaining 971 non-progressing workflows simply sit there waiting for their jobs to run. However, all 1,000 workflows are marked “Running” in the Terra UI, so you have no way to know what to expect and will quickly start worrying that something is wrong.
In order to provide more clarity in such cases, we introduced an enhanced workflow starting algorithm in Cromwell that takes into account whether an incoming workflow will be able to run jobs or not. If Cromwell determines that a workflow will not be able to start due to job capacity limitations, it holds the workflow in a separate queue with a clear “Queued in Cromwell” status indicator until capacity becomes available.
Start workflows from low-volume users first
Finally, we turned to look at how we were balancing the relative needs of researchers and groups across a range of use cases — from the individual WDL developer who submits one workflow at a time and needs to iterate quickly, to the genome center project manager who submits thousands of samples at a time and needs execution to proceed reliably, with minimal intervention.
Until now, Cromwell did not apply any prioritization in how it handled incoming workflows; it was just first-come, first-served. As a result, it would happily process a 100,000 workflow submission in its entirety before moving on to a 10-workflow development submission. That is actually where Rawls’ quota of 3,000 workflows per user came from — it was a way to ensure a hard maximum for how long anyone might be waiting behind another submission.
To address this more elegantly, we implemented a prioritization algorithm designed to minimize latency for small users, while preserving the high capacity demanded by large users:
- Identify all projects that have workflows wanting to run
- Sort projects by the lowest number of currently-running workflows
- Start 1 workflow from that group
- When groups are tied, select the group that started a workflow least-recently
- Wait for next clock tick
- Goto (1)
This is an especially big deal for small users, since there is no longer a scenario where they have to wait for the genome center’s thousands of workflows to get out of the way. Meanwhile, large users will barely notice because small users are… small. And in the event of multiple large users launching large submissions at the same time, the algorithm will elegantly bring their workflow counts into stable equilibrium.
In the future —pending additional testing— we hope to capitalize on the new, smarter Cromwell to significantly raise Rawls’ quota for concurrent workflows per user. This will enable Cromwell to operate closer to full, optimal capacity, without the need for Rawls to play referee.
