Dr. Kiran Garimella is the Associate Director for Genomic Medicine in the Data Sciences Platform at the Broad Institute. In this guest blog post, Kiran gives us an overview of MAS-ISO-seq: a new method for high-throughput long-read transcriptome sequencing, collaboratively developed with Dr. Aziz Al’Khafaji (Hacohen/Blainey labs); Jonn Smith (DSP), Dr. Mehrtash Babadi (DSP), and the help and support of many others at the Broad Institute.
You can see the data processing part of the MAS-ISO-seq method in action in the public Terra workspace created by the development team, which features a workflow going from raw PacBio output to properly segmented and filtered reads ready for analysis, configured to run on a publicly accessible example dataset.
Short reads (~150-200 bp) are the standard brush for painting a picture of gene expression activity. But long read bristles are much finer, revealing more detail than possible with short reads alone. PacBio instruments are capable of generating long (~15,000-25,000 bp) and accurate (~Q30) reads via circular consensus sequencing (aka “HiFi”). As the average gene transcript isoform lengths are only ~1,500 bp, most full-length isoforms are easily capturable. Long reads enable identification of alternatively/aberrantly spliced isoforms and gene fusions, comparison across samples and cell types, all without the need for de novo assembly or other complex reconstruction methodologies.
The downside, however, is yield: long read output is comparatively low, limiting their scalability and applicability to interesting biological problems.
On PacBio’s long read sequencing platforms specifically, yield is dictated by the number of active nanoscopic wells on each flowcell. The current flowcell design has around 3 to 6 million active wells, which generates an equal number of reads regardless of their length: you only get one read out per well, whether the transcript is 1,500 bp or 25,000 bp long.
So here’s the big question: if the instrument can sequence such long stretches of DNA, and cDNA sequences are much shorter than that typical length, is there some way to overcome the one-read-per-well limitation and get much more data from each flowcell?
Yes! By applying a new within-read multiplexing approach to PacBio sequencing, we were able to develop a new protocol that increases the yield of full-length RNA isoform sequence data by 15x. The protocol is called Multiplexed ArrayS isoform sequencing, or MAS-ISO-seq, and is suitable for both single-cell and bulk sequencing applications.
You can find the full details of how it works in our preprint in bioRxiv, or keep reading to get the highlights from this blog.
Using intramolecular multiplexing to maximize yield
The central idea of this approach is to combine several cDNA sequences into a single molecule (which we term an “array”), sequence that, then segment the resulting long read into its constituent fragments for analysis.
To do this, we start with a cDNA library (usually a single cell library that has been prepared with a 10x Genomics kit) and split it up among several PCR reactions. In each PCR reaction, we add a different barcode pair. What’s really neat is that the barcode pairs are designed to be complementary across parallel reactions, so when we process the ends to make them “sticky” and pool them all together, the barcoded cDNA fragments self-assemble into the array.
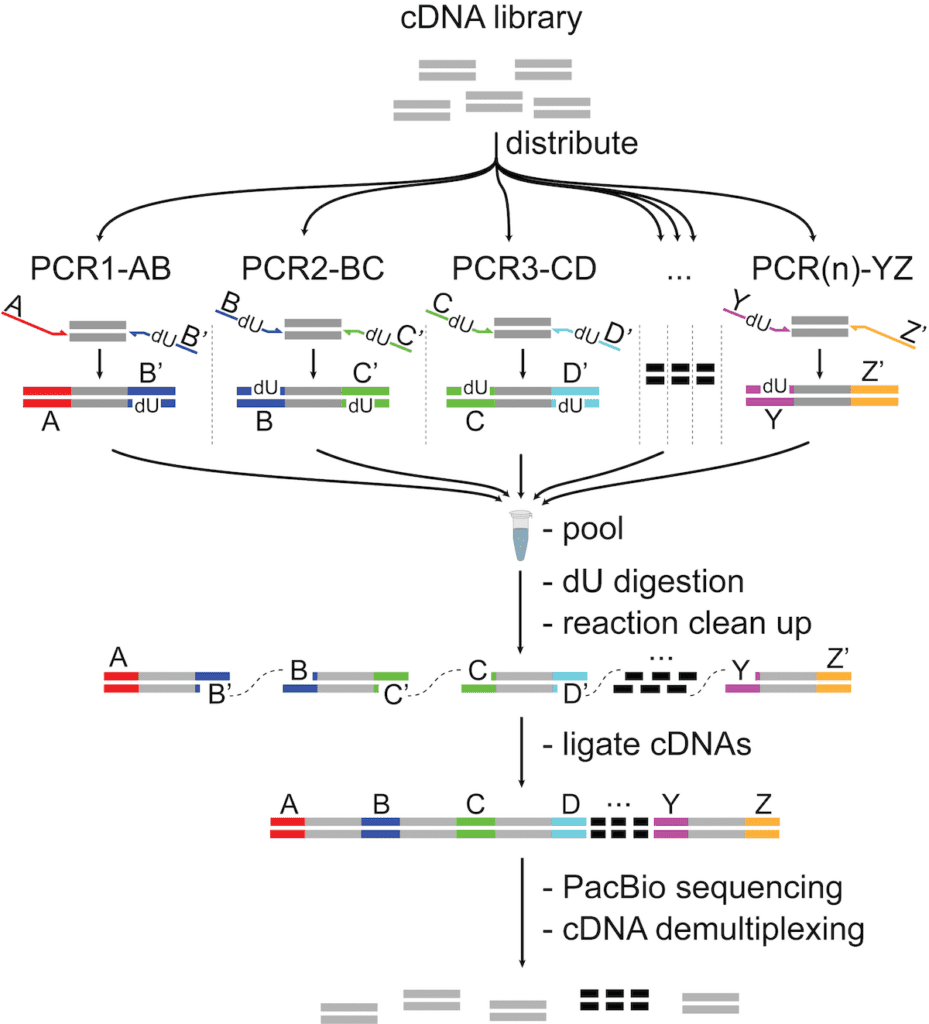
Schematic of the MAS-ISO-seq intramolecular cDNA multiplexing workflow
The barcodes in the array have a predictable, programmatic order (A, B, C, D, E, … etc.), and the maximum number of cDNA sequences in the read is constrained by the number of parallel PCR reactions (we usually do 15, which works out to a 22,500 bp read if every transcript is 1,500 bp).
A probabilistic approach to read segmentation
Once we’ve sequenced each multiplexed array, we need to split the resulting long read up into the individual sequences corresponding to the original cDNA fragments.
We initially tried using an iterative BLAST-like search to find the adapters and split the read at those positions, but that approach turned out to be quite brittle. It doesn’t understand context (“does it make sense that this substring in the read is a barcode given what’s surrounding it?”) and it’s extremely sensitive to sequencing error (“this read wasn’t error corrected as well as the last one; can I really believe that the subsequence I’m looking at is one of my barcodes?”). This is a big problem: mis-segmented reads may be misleading, masquerading for instance as false fusion genes or other seemingly interesting —but wrong— biological findings.
The quality control evaluation of our initial pilot run confirmed that the BLAST-like adapter search was not going to cut it (so to speak) so we pivoted to a different solution that leverages what we know about the structure of our MAS-ISO-seq reads:
- Array elements (i.e. a transcript with poly-A tail and optional single cell adapters, etc.) are flanked by our barcodes, referred to here as Mi and Mi+1. Across the length of the read, these adapters appear as an ordered sequence (M1, M2, M3, …, Mn) where n is the total number of elements in the array.
- Each array element itself contains several sequences known a priori (e.g. 10x Genomics single-cell 5’ and 3’ adapters, poly-A tails).
- The cDNA sequence itself can be considered not known a priori. Cell barcodes, spatial barcodes, UMIs, or other adapters that come from a very large sequence space may also effectively be considered unknown.
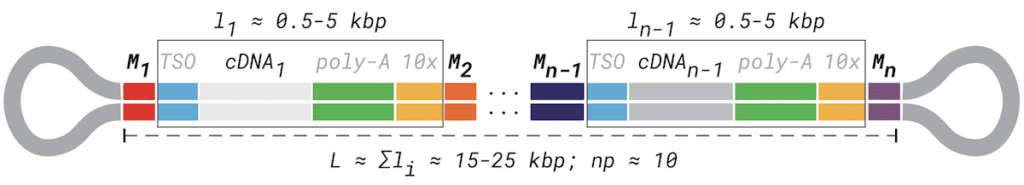
MAS-ISO-seq read structure barcodes and other adapters arising from the single-cell library preparation additionally highlighted.
This codified read structure lays out useful landmarks and constraints, which enabled us to develop a model for annotating then segmenting them with high fidelity, even in the presence of high sequencing error rates.
In particular, the latter two observations enabled us to design a composite hidden Markov model with two separate submodels: “global alignment” and “random, repeat”. The “global alignment” submodel enables the recognition of known sequences (not just the MAS-ISO-seq barcodes, but all of them), allowing for mismatches and indels along the length of the sequence. The “random, repeat” submodel enables the recognition of sequences that are not known in advance. These models are connected to one another and repeated as necessary according to a given MAS-ISO-seq array design (for example, a 15-element array). See the preprint if you want full technicolor detail (and some nifty grayscale figures) about the submodels; the point is that this modeling approach gives us much higher confidence in our annotation of where the sequences of interest start and stop within each array read.
With the annotated reads in hand, we can apply the ordering constraint outlined in the first observation as a quality control check. Based on the patterns recorded during the annotation stage, we can confirm whether each array read’s MAS-ISO-seq adapters appear in the order specified by the array design. Reads failing this check are potentially mis-segmented, so we filter them out. And that’s it!
We implemented this method as an open-source software package called Longbow, which is available for download on Github and through Pypi, and we also provide a workflow script along with example data in a Terra workspace (see Resources at the end of this post for details).
A >15x increase in single-cell full-length transcript isoform yield
To evaluate the performance of our approach on a full-size dataset, we applied Longbow to 15-element MAS-ISO-seq data for a test dataset on T-cells. We started with an initial set of ~5.6M input reads, of which ~1.6M reads were successfully error-corrected to ~Q30 by PacBio’s on-board software. Of these error-corrected reads, ~99% were successfully demultiplexed into 22.7M CCS-corrected transcripts, a ~14x increase from the initial 1.6M corrected read set.
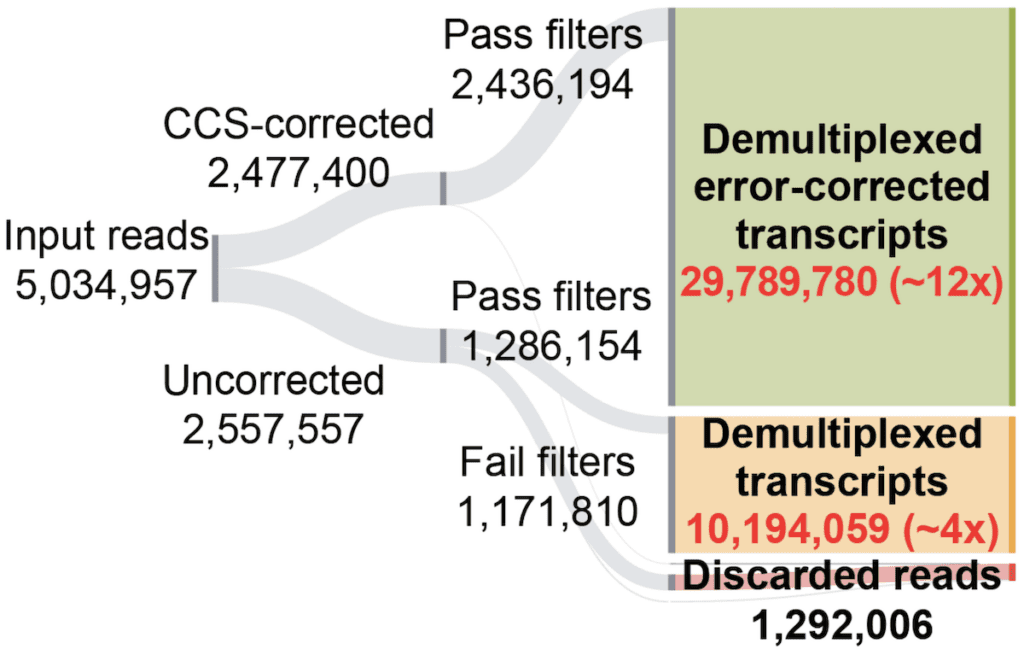
Sankey diagram of final processing status for reads from an exemplar 15-element MAS-ISO-seq library
What happened to the other 3.9M reads, you ask? Well, usually those get thrown away. PacBio’s error correction protocol relies on molecules being redundantly sequenced within each well and then transformed into a consensus sequence. Many reads, however, don’t get enough redundant passes to be corrected, and are therefore discarded.
But uncorrected doesn’t mean unusable; we can rescue this data! Because our HMM is robust to sequencing error, we also process the ~3.9M reads that PacBio’s software does not correct. Here, substantially more reads fail Longbow’s filtering (~55%). However, 1.7M reads do segment properly and pass filtration, yielding another ~12.8M transcripts (additional ~8x increase from the initial 1.6M corrected reads). These transcripts are not error-corrected, but still represent valid data that can be used for a variety of purposes (e.g. transcriptome annotation).
More power to identify cell types and differential expression
What can we do with the massive increase in full-length transcriptome data enabled by MAS-ISO-seq? Let’s look at a single single-cell library on ~6,000 tumor-infiltrating cytotoxic T cells, prepared both for short read sequencing and as a MAS-ISO-seq library. The figure below shows our short read dataset, with cells clearly separated into clusters representing different states of dormancy/activation/exhaustion (panel A).
When we downsample our MAS-ISO-seq output to different levels (1.6M to 33M reads, panel B), we can see how more long read data influences a similar clustering analysis. As we add more data, cell clustering becomes more stable, increasingly resembling that of the short reads.
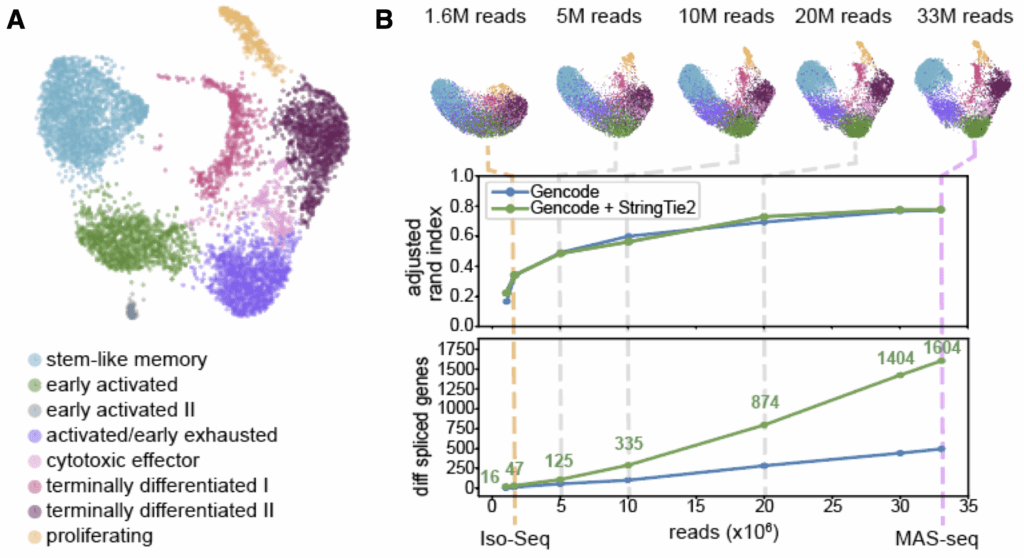
Comparison of short read clustering to in silico downsampling of long read data. (A) UMAP embedding of single-cell gene expression of 6,260 CD8+ T cells from short (B) In silico downsampling analysis of MAS-ISO-seq reads; (top) evolution of UMAP embedding vs. depth (the long-read UMAPs are annotated with the cell identities determined from the short-read data); (middle) adjusted Rand index (ARI) between short-reads reference annotations and downsampled long reads vs. depth; (bottom) number of statistically significant differentially spliced genes vs. depth.
Adding more data also allows us to do progressively better at annotating a custom transcriptome for these cells – using StringTie2 to augment the canonical Gencode annotations with high-confidence transcripts found in these cells. Our ability to identify differentially spliced genes among cell clusters increases substantially as a function of overall read count: up to a 34-fold gain (1604 vs 47) when we use the entire dataset!
Future directions and availability of the software
We’re very excited by MAS-ISO-seq’s ability to produce substantially higher throughput full-length isoform sequencing in single cells. And this is just the start! Over the coming months, we plan to add more capabilities to our processing software, Longbow. Support for more array designs (e.g. 10x Genomics 3’ kit, spatial transcriptomics, etc.), support for full-length poly(A) tail capture, more robust training for the HMM, speed and usability enhancements, and more.
Like all software authored in the Data Sciences Platform, Longbow is open-source (BSD-3-Clause License) and should work on any reasonable HPC environment. We also provide a Docker image with all of the relevant dependencies preinstalled, as well as an easy-to-install PyPi package.
For your convenience, we also provide a reproducible workflow for going from raw PacBio output to properly segmented and filtered reads ready for analysis. We plan to make further improvements to this workflow in the coming months, such as the ability to output gene-level and isoform-level quantification matrices that can be imported directly into Scanpy for analysis. The workflow is available from Github and Dockstore, and we make it available in a public Terra workspace, preconfigured to run on some example data, to enable anyone to try out the method quickly without any setup work. Please check it out and let us know what you think!
Resources
- The MAS-ISO-seq preprint can be found at https://www.biorxiv.org/content/10.1101/2021.10.01.462818v1. Comments/reviews are most welcome!
- The Longbow software can be found at https://github.com/broadinstitute/longbow and on Pypi at https://pypi.org/project/maslongbow/, with documentation at https://broadinstitute.github.io/longbow/. Comments and issue reports are welcome there too.
- An example dataset of Spike-In RNA Variants (Lexogen SIRV-Set 4) and the workflow for initial data processing can be found in this Terra workspace.
- If you are new to running workflows in Terra, see the Workflows Quickstart Tutorial to learn how you can use the preconfigured workspace to try out the method yourself.
